Sara Rosenbaum's Work
We found 35 items
Include in results:
#1
 96694.5
96694.5
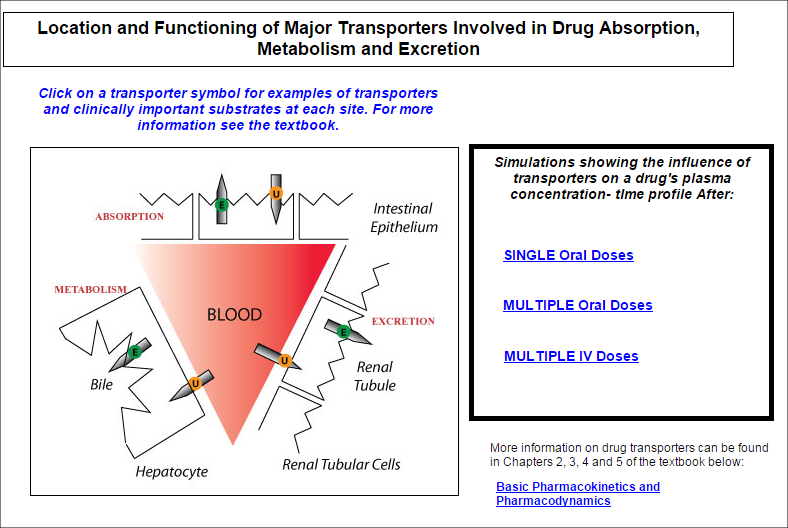
Model 2. Drug Transporters
This simulation shows the location and functioning of the major clinically important drug transporte
... Read more
This simulation shows the location and functioning of the major clinically important drug transporters in the : gastrointestinal membrane; renal tubular membrane; and the membranes of the hepatocyte. Simulations demonstrate their functioning after oral and intravenous doses of a drug substrate
sim
⋮ 73,592 runs
drug
transporters
efflux
uptake
drug absorption
#2
 91434.02
91434.02
Model 1. Introduction to PK and PD
This model allows users to use trial and error to identify appropriate doses of a drug for IV, oral
... Read more
This model allows users to use trial and error to identify appropriate doses of a drug for IV, oral and other extravascular routes of administration
sim
⋮ 104,443 runs
Pharmacokinetics
Pharmacodynamics
Therapeutic Range
#3
 86045.77
86045.77
Intravenous Infusion
This model demonstrates the pharmacokinetic characteristics of constant continuous drug administrati
... Read more
This model demonstrates the pharmacokinetic characteristics of constant continuous drug administration.
sim
⋮ 43,547 runs,
316 downloads
Download Model
Download Model
drug
pharmacokinetics
intravenous infusion
zero order input
constant continuous drug administration
#4
 69977.52
69977.52
Oral Absorption
This model is based on first order absoprtion in a 1-compartment model
#5
 59166.7
59166.7
Mulitple Bolus Injections Pharmacokinetics
This model shows the typical plasma concentration profile associated with the administration of seve
... Read more
This model shows the typical plasma concentration profile associated with the administration of several doses of a drug over time. It demonstrates the determinants of the fluctuation in the plasma concentrations, the accumulation of the drug over the course of therapy and the resting steady state plasma concentrations.
sim
⋮ 35,572 runs,
243 downloads
Download Model
Download Model
drug
pharmacokinetics
multiple doses
multiple intravenous bolus injections
#6
 40952.15
40952.15
Multiple Oral Doses Pharmacokinetics
Most commonly people take oral doses of a drug over an extended period. This model demonstrates the
... Read more
Most commonly people take oral doses of a drug over an extended period. This model demonstrates the unique plasma concentration profile associated with this type of drug administration. The model assumes first-order drug absorption with no lag time. Simulations can be carried out to observe how the rate and extent of absorption (bioavailability) affect the profile.
sim
⋮ 25,842 runs,
204 downloads
Download Model
Download Model
multiple oral doses
interactive pharmacokinetics
drug
bioavailability
#7
 31169.3
31169.3
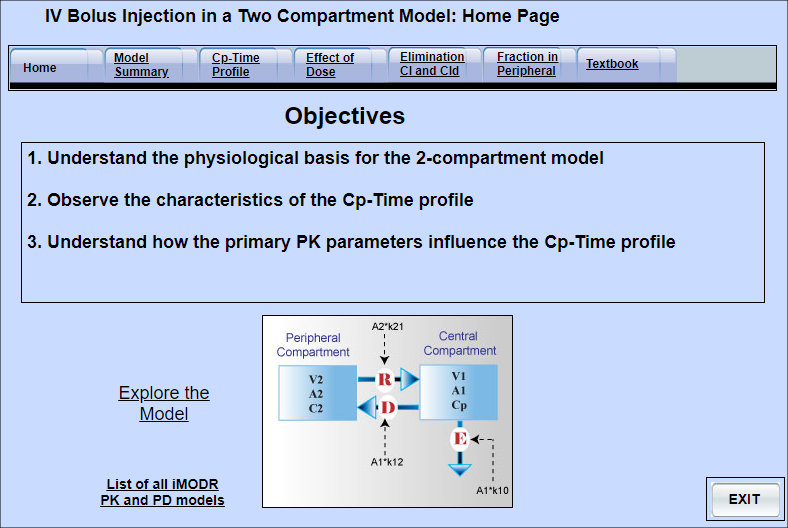
Pharmacokinetics: An IV Injection on a Two Compartment Model
This model shows the profile of a drug after an intravenous injection when the drug displays two com
... Read more
This model shows the profile of a drug after an intravenous injection when the drug displays two compartment characteristics
#8
 24859.553
24859.553
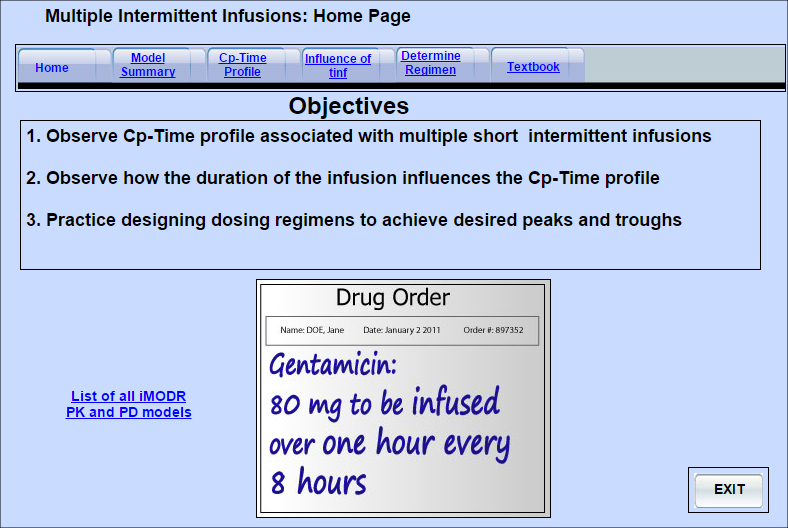
Multiple Intermittent Infusions Pharmacokinetics
The model shows the unique plasma concentration-time profile associated with the administration of a
... Read more
The model shows the unique plasma concentration-time profile associated with the administration of a drug using multiple short infusions. It demonstrates the influence of the duration of the infusion and allows the user to practice the calculation of a suitable dose and dosing interval to achieve desired peak and trough plasma concentrations of a drug.
sim
⋮ 12,438 runs,
165 downloads
Download Model
Download Model
Multiple intermittent infusions
aminoglycoside infusion
drug
interactive pharmacokinetics
#9
 22089.541
22089.541
Understanding Restrictive and Nonrestrictive Hepatic Clearance
Many drugs are removed from the body by metabolism in the liver. A drug's hepatic clearance is a me
... Read more
Many drugs are removed from the body by metabolism in the liver. A drug's hepatic clearance is a measure of the liver's ability to metabolize a drug. This simulation demonstrates the characteristics of high extraction (nonrestrictive clearance, low extraction (restrictive clearance and intermediate clearance.
sim
⋮ 11,635 runs
drug
metabolism
clearance
elimination
restrictive clearance
#10
 20997.56
20997.56
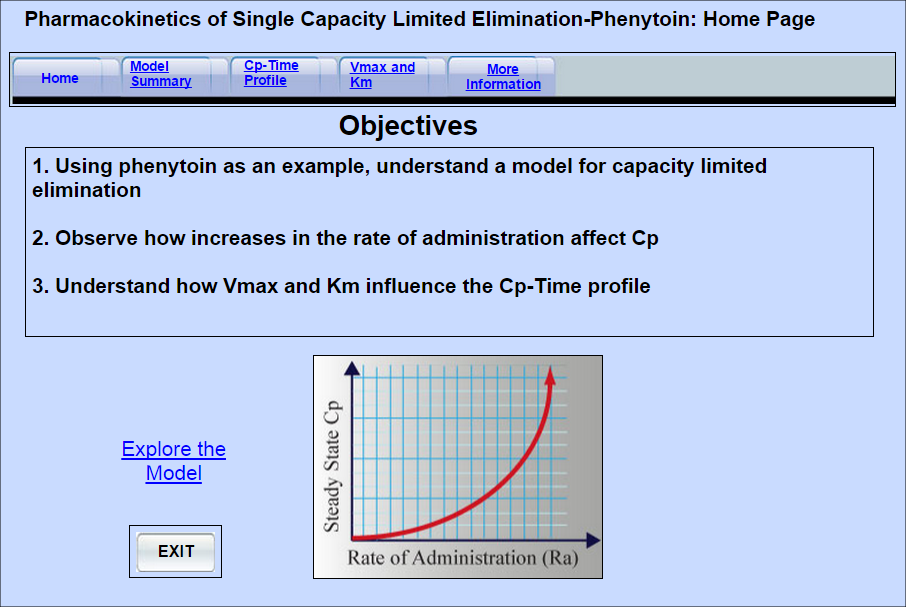
Nonlinear Pharmacokinetics and Phenytoin
Nonlinear or capacity limited pharmacokinetics can occur whenever a process involved in the absorpti
... Read more
Nonlinear or capacity limited pharmacokinetics can occur whenever a process involved in the absorption, distribution or elimination of a drug becomes saturated. This model demonstrates nonlinear elimination (metabolism) using phenytoin as the model drug. Specifically, it demonstrates how increasing doses of the drug produce disproportionate increases in the plasma concentration. It also demonstrates how the model parameters, Km and Vmax influence of the plasma concentration -time profile.
sim
⋮ 13,178 runs,
173 downloads
Download Model
Download Model
nonlinear pharmacokinetics
capacity limited elimination
drug
phenytoin
#11
 18526.826
18526.826
Take the Infusion Challenge
Use this model to see if you can use a drug's pharmacokinetic and pharmacodynamic properties to dete
... Read more
Use this model to see if you can use a drug's pharmacokinetic and pharmacodynamic properties to determine the initial rate of drug administration. See if you can identify potential drug-drug interactions and respond when appropriate by making appropriate modifications to the dose.
sim
⋮ 20,399 runs
Dosing
Drug
Pharmacokinetics
Constant Continuous Input
#12
 15206.88
15206.88
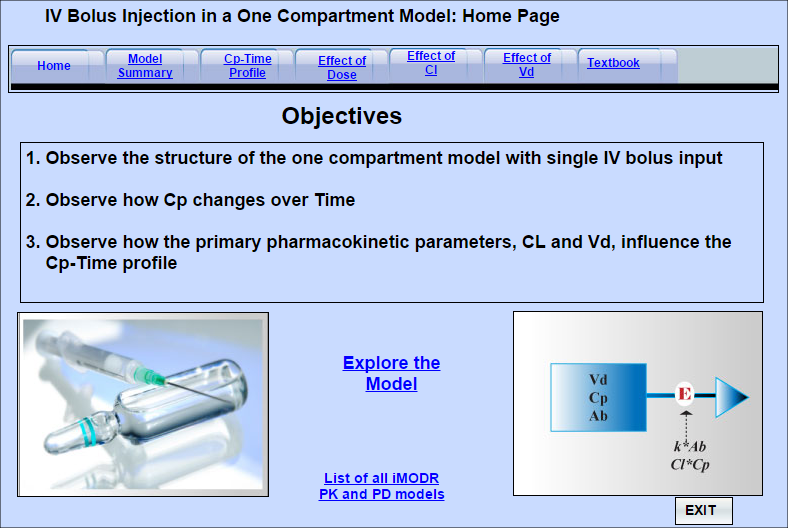
Pharmacokinetics: An IV Injection in a 1-Compartment Model
#13
 13805.023
13805.023
Sigmoidal Emax Model of Drug Response
This is the model that is most commonly used to for the time course of drug effects in man. It assum
... Read more
This is the model that is most commonly used to for the time course of drug effects in man. It assumes that the response is driven by the blood or plasma concentrations of the drug. When the model parameters are known, it can be used to predict response at anytime time after any dose.
#14
 10795.228
10795.228
Tolerance-Nicotine- Hypothetical Antagonist
Tolerance may be defined as a process that results in a reduction in the response to a specific drug
... Read more
Tolerance may be defined as a process that results in a reduction in the response to a specific drug concentration following repeated drug exposure. One model for tolerance assumes that the drug produces a hypothetical metabolite that opposes its action.
#15
 8054.796
8054.796
Model 15. DDI 3 Reversisble Enzyme Inhibition: Midazolam Fluconazole
This model demonstrates the characteristics of reversible enzyme inhibition using fluconazole's inhi
... Read more
This model demonstrates the characteristics of reversible enzyme inhibition using fluconazole's inhibition of midazolam as an example.
sim
⋮ 5,099 runs
Reversible inhibition
Competitive inhibition
Midazolam
Fluconazole
Enzyme inhibition
#16
 7594.4375
7594.4375
Model 20. DDI Drug-Drug Interactions - Pharmacodynamic
This model demonstrates the interaction between two agonists. The pharmacodynamic characteristics of
... Read more
This model demonstrates the interaction between two agonists. The pharmacodynamic characteristics of the perpetrator drug can be changed to create a full agonist, a partial agonist and a full antagonist
sim
⋮ 4,875 runs
DDI
Drug Drug Interactions
Pharmacodynamic
Full agonist
Partial agonist
#17
 5754.573
5754.573
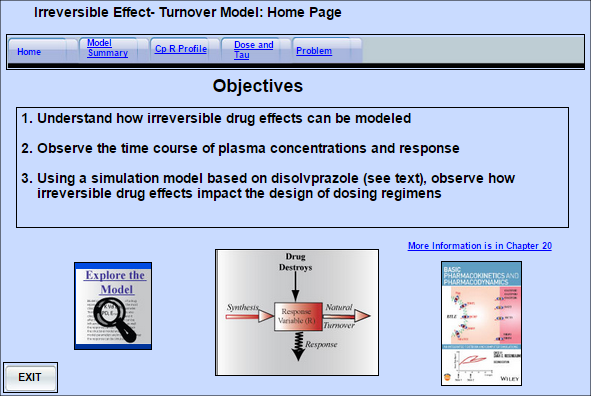
Irreversible Drug Effects - Proton Pump Inhibitors
Some drugs act by binding covalently to their receptors. As a result, the target is destroyed and it
... Read more
Some drugs act by binding covalently to their receptors. As a result, the target is destroyed and its function returns only when it has been replaced by newly synthesized product. The target may be a protein, DNA, an enzyme, or a cell at any stage of development. This model has been applied to the action of the proton pump inhibitors, which bind to and destroy the H+,K+-ATPase pumps in the parietal cells of the gastric mucosa. Normal proton secretion is restored only when the pumps are replaced by newly synthesized functioning pumps (i.e., the usual turnover time of the system).
sim
⋮ 2,836 runs,
148 downloads
Download Model
Download Model
Pharmacodynamic
Irreversible Drug Effects
Proton Pump Inhibitors
PPI Response Model
#18
 5006.014
5006.014
Physiologically Based Pharmacokinetic Model
This model shows how to build a PBPK model and how drug concentrations in different tissues can be s
... Read more
This model shows how to build a PBPK model and how drug concentrations in different tissues can be simulated
sim
⋮ 3,765 runs
Drug
Pharmacokinetic
PBPK
Physiologically Based Pharmacokinetic Model
#19
 4371.596
4371.596
Model 21. Emax Model of Drug Response With an Effect Compartment
This model demonstrates how an effect compartment can be added to a pharmacokinetic model to accommo
... Read more
This model demonstrates how an effect compartment can be added to a pharmacokinetic model to accommodate a delay in drug response caused by a slow distribution of a drug to its site of action. The simulation also explains hysteresis
#20
 3732.51
3732.51
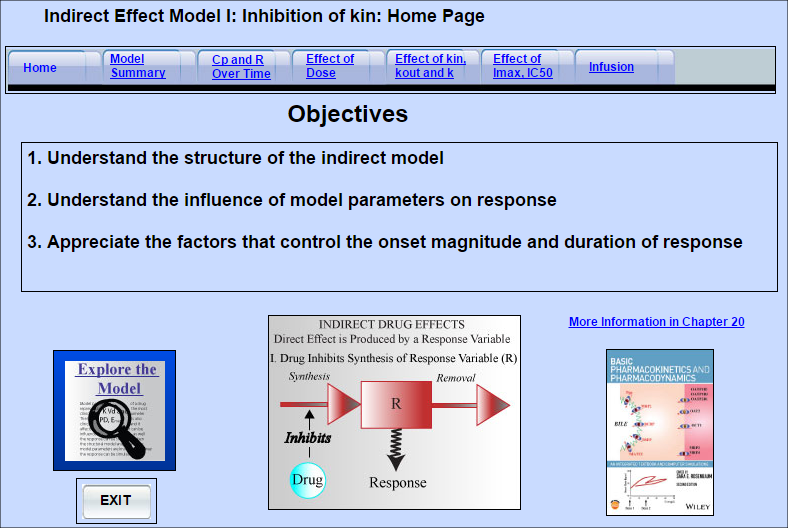
Indirect Effect Model 1
Some drugs do not directly produce the measured drug response. Instead they act upstream, and either
... Read more
Some drugs do not directly produce the measured drug response. Instead they act upstream, and either increase or decrease the amount of the entity that directly mediates the response (response variable). Indirect effect Model I can be used for drugs that inhibit the synthesis of the response variable. An example is warfarin which inhibits the synthesis of clotting factors.
#21
 2697.25
2697.25
Transit Compartment of Drug Action
Change image
Submit your own comment on the simulation.
Share Comment
upgrade hosting plan d
... Read more
Change image
Submit your own comment on the simulation.
Share Comment
upgrade hosting plan delete simulation
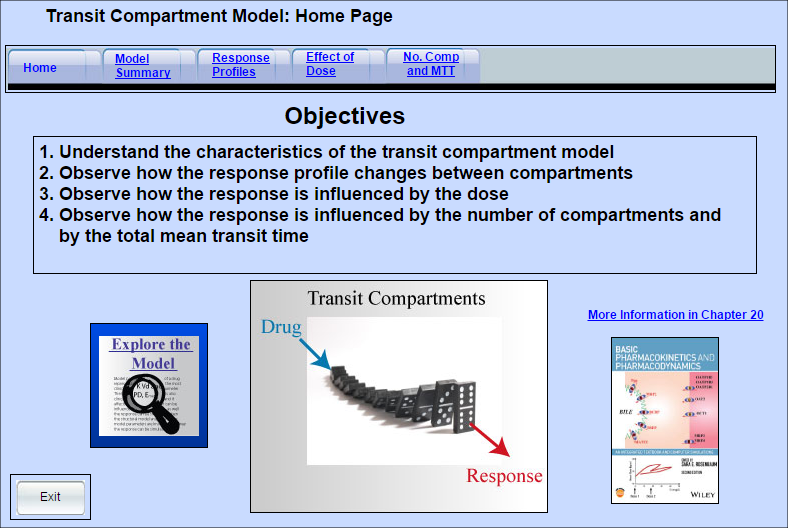
Transit Compartment Model of Drug Response
By Sara Rosenbaum
Sim URL: https://forio.com/simulate/sarar/transit-compartment-model
Sim access:Other authors can download source model
Sim plan: Simulate Free
Sim stats:This sim has been run 326 times.
This simulation was uploaded to Forio Simulate with the isee NetSim software. More information can be found at iseesystems.com.
Your Rating:
1 star2 star3 star4 star5 star
Average Rating:
rating(1)
Click here to edit the description
A delay in response to a drug can occur when it takes a long time for the drug’s initial effect to be translated into the final response (a long transduction process). The delayed response profile can be captured using a series of transit compartments.
sim
⋮ 1,490 runs,
157 downloads
Download Model
Download Model
Pharmacodynamics
Transit compartment
Delayed drug action
#22
 2473.1536
2473.1536
Model 20. DDI Drug-Drug Interactions - Pharmacodynamic
This model demonstrates the interaction between two agonists and allows the efficacy (Emax) of one t
... Read more
This model demonstrates the interaction between two agonists and allows the efficacy (Emax) of one to be varied
#23
 1880.135
1880.135
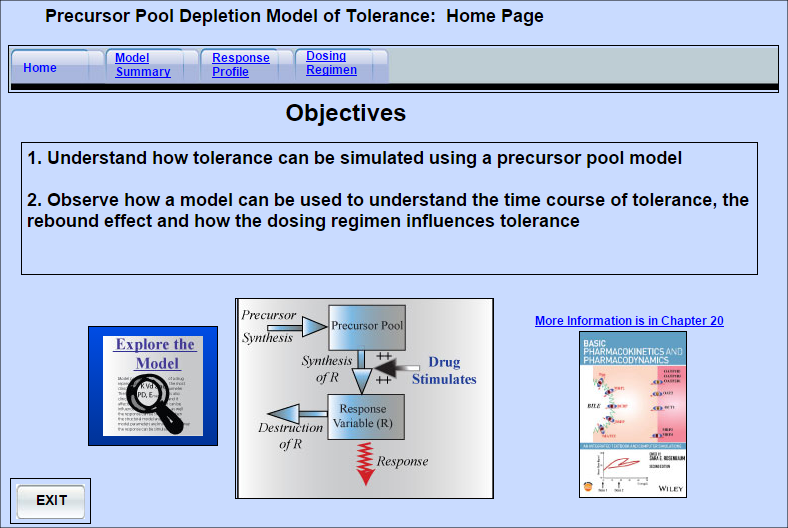
Tolerance- Precursor Pool Model
Tolerance may be defined as a process that results in a reduction in the response to a specific drug
... Read more
Tolerance may be defined as a process that results in a reduction in the response to a specific drug concentration following repeated drug exposure. Tolerance could occur if an endogenous compound that plays an essential role in the response chain becomes depleted during response. This model assumes the drug stimulates the production of a response variable which becomes then becomes depleted.
sim
⋮ 1,268 runs,
133 downloads
Download Model
Download Model
Tolerance
Pharmacodynamic
precurosr pool depletion model
#24
 1851.6566
1851.6566
Indirect Effect Model 4
Some drugs do not directly produce the measured drug response. Instead they act upstream, and either
... Read more
Some drugs do not directly produce the measured drug response. Instead they act upstream, and either increase or decrease the amount of the entity that directly mediates the response (response variable). Indirect effect Model IV can be used for drugs that stimulate the degradation of the response variable. As a result they decrease the amount of the response variable.
sim
⋮ 1,382 runs,
134 downloads
Download Model
Download Model
Drug
Pharmacodynamics
Indirect Effect Model 4
Stimulation of kout
#25
 1826.996
1826.996
Extended Hepatic Cl Model
This model assumes permeability controlled hepatic uptake and demonstrates the role of passive diffu
... Read more
This model assumes permeability controlled hepatic uptake and demonstrates the role of passive diffusion, hepatic uptake transporters, hepatic matabolism and hepatic efflux transporters on overall hepatic elimination
sim
⋮ 816 runs
Biology
Science
Education
Extended clearance
Permeability controlled distribution
#26
 1553.1493
1553.1493
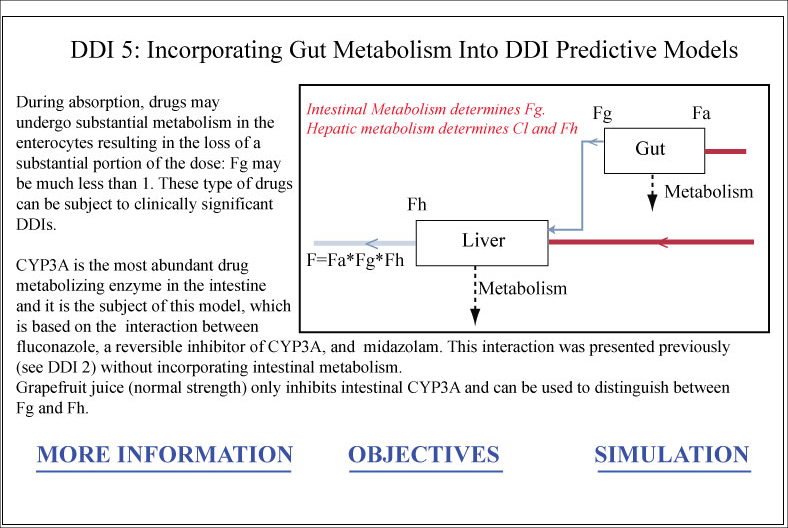
Model 17. DDI 5 Inhibition of Gut and Hepatic Metabolism
This model demonstrates the effect of an inhibiotr that acts on enzymes both the gastrointestinal me
... Read more
This model demonstrates the effect of an inhibiotr that acts on enzymes both the gastrointestinal membrane and the liver. It also demonstrates the effect of grapefruit juice on drugs netaboliized by CYP3A
sim
⋮ 857 runs
Intestinal extraction
Fg
DDI
Drug interactions
Grapefruit juice
#27
 1428.2587
1428.2587
Model 34. Fentanyl Induced Respiratory Depression and its Reversal With Naloxone
This models demonstrates the varying degrees of respiratory depression caused by fentanyl and its re
... Read more
This models demonstrates the varying degrees of respiratory depression caused by fentanyl and its reversal by naloxone. It also demonstrates that the action of naloxone is short lived and that additional doses may be needed
sim
⋮ 2,085 runs
Opioid
opioid overdose
naloxone
Narcan
#28
 1187.7
1187.7
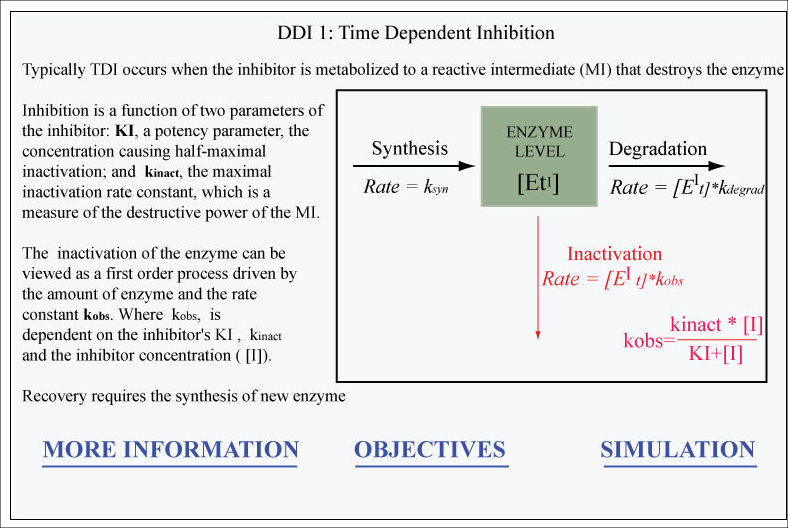
Model 13. DDI 1 Time Dependent Enzyme Inhibition
This simulation demonstrates the properties of time dependent enzyme inhibition. Specifically it sho
... Read more
This simulation demonstrates the properties of time dependent enzyme inhibition. Specifically it shows how the inhibitor’s KI and kinact and the derived parameter kobs, control inhibition. It also demonstrates how the enzyme’s degradation rate constant controls recovery.
sim
⋮ 810 runs
Time dependent enzyme inhibition
mechansim based inhibition
recovery
#29
 1077.1764
1077.1764
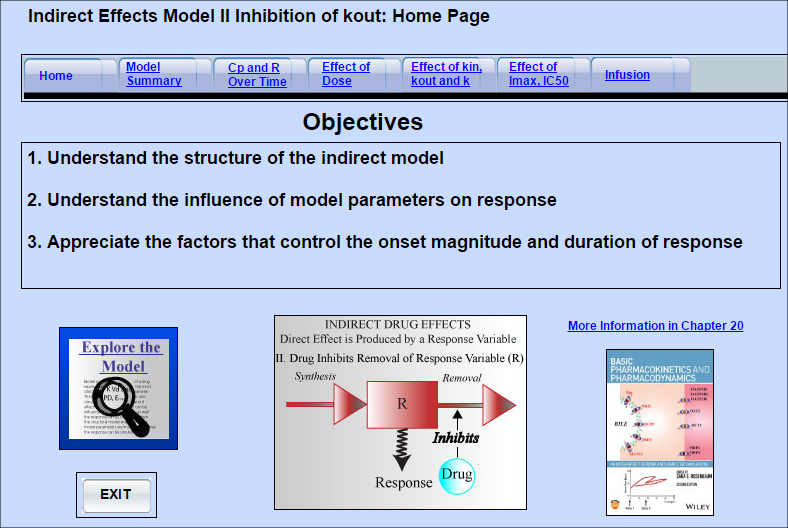
Indirect Effect Model 2
Some drugs do not directly produce the measured drug response. Instead they act upstream, and either
... Read more
Some drugs do not directly produce the measured drug response. Instead they act upstream, and either increase or decrease the amount of the entity that directly mediates the response (response variable). Indirect effect Model 2 can be used for drugs that inhibit the degradation of the response variable. As a result they increase the amount of the response variable.
#30
 926.62134
926.62134
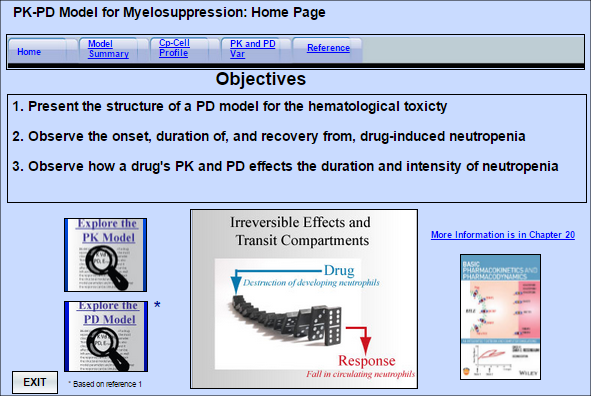
Model 27. Hematological Toxicity of Anticancer Drugs
This model shows the typical effect of several anticancer drugs on the number of circulating neutrop
... Read more
This model shows the typical effect of several anticancer drugs on the number of circulating neutrophils. The drugs destroy neutrophils as they develop in the bone marrow, and its takes several days for their action to affect the circulating neutrophils. The model demonstrates how inter-individual variability in pharmacokinetics and pharmacodynamics can effect the magnitude and duration of this response.
#31
 855.28735
855.28735
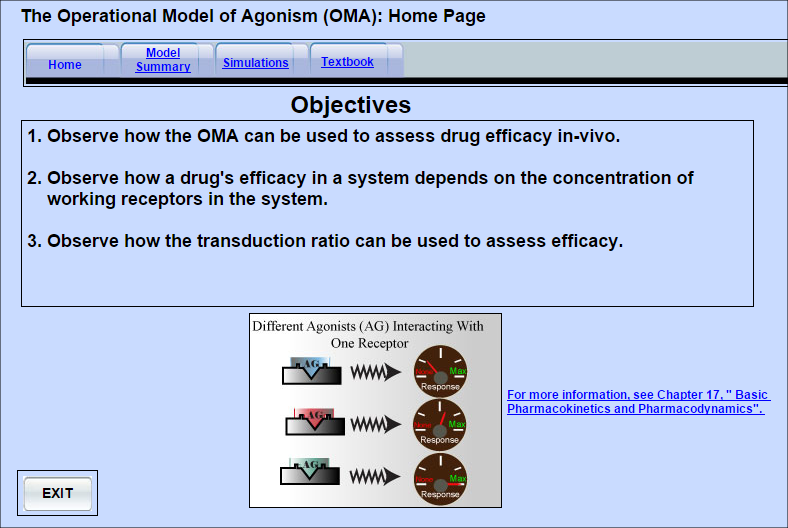
Operational Model of Agonism
The operational model of agonism provides a way of measuring drug efficacy and affinity in-vivo.
sim
⋮ 2,775 runs
Pharmacodynamics
#32
 700.508
700.508
Model 14. DDI 2 Enzyme Induction
This simulation demonstrates the model used for the induction of the drug metabolizing enzymes. It i
... Read more
This simulation demonstrates the model used for the induction of the drug metabolizing enzymes. It illustrates how the inducing drug's characteristics (Emax and EC50) control the degree of induction and the rate of degradation of the enzyme controls recove..
sim
⋮ 708 runs
pharmacokinetics
enzyme induction
rifampin
#33
 576.865
576.865
Integrated PK-PD Model For Lipoamide
Lipoamide is a fictitious antipyretic (fever reducing) drug that is believed to work by reducing the
... Read more
Lipoamide is a fictitious antipyretic (fever reducing) drug that is believed to work by reducing the synthesis of cytokines. Based on its mechanism of action and the characteristics of its response, an indirect effect model I (inhibition of kin) was used to model its effect. This model can be used to probe optimum dosing regimens of lipoamide.
sim
⋮ 497 runs
Indirect effect model
Inhibition of kin
pharmacodynamics
#34
 567.1
567.1
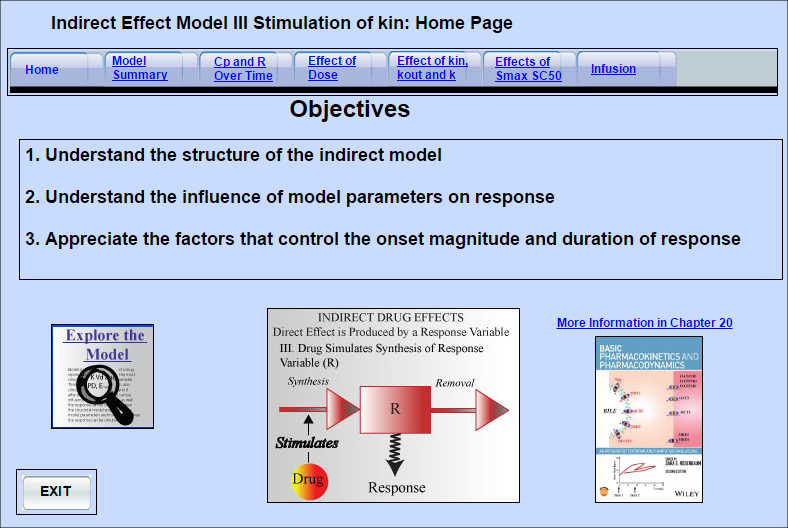
Indirect Effect Model 3
Some drugs do not directly produce the measured drug response. Instead they act upstream, and either
... Read more
Some drugs do not directly produce the measured drug response. Instead they act upstream, and either increase or decrease the amount of the entity that directly mediates the response (response variable). Indirect effect Model III can be used for drugs that stimulate the production of the response variable. As a result they increase the amount of the response variable.
sim
⋮ 335 runs,
133 downloads
Download Model
Download Model
Drug
Pharmacodynamics
Indirect Effect Model 3
Stimulation of kin
#35
 377.311
377.311
Model 16. DDI 4 Time Dependent Enzyme Inhibition Desipramine and Paroxetine
This simulation demonstrates the characteristics of time dependent enzyme inhibition using paroxetin
... Read more
This simulation demonstrates the characteristics of time dependent enzyme inhibition using paroxetine's inhibition of desipramine as an example
sim
⋮ 262 runs
DDI
Drug interactions
Time dependent inhibition
Paroxetine
Desipramine
Uploading a Bundle from Zip
Instead of creating bundles, categories, and assemblies one by one, you can upload a single zip file that contains all of your bundle's content. To create your zipped bundle, make a folder with your bundle's name and add subfolders with your categories' names. The folder tree should have the same structure that you want the categories to have in your bundle. Place your assembly .stmx files in the appropriate category folders, then zip your bundle folder and upload it using the Upload Bundle from Zip link above.
Assemblies, Bundles, and Categories
Assemblies are self contained models that demonstrate common ways to connect together building blocks and that can be used as parts of other models. This is analogous to using prefabricated wall and roof pieces to construct a house.
Bundles are groups of assemblies with a common use or theme. For example, a Health Care bundle might contain a variety of assemblies that aid in creating health care models. When you download assemblies from the isee Exchange™, you download an entire bundle, rather than individual assemblies.
Categories are subgroups of assemblies within a bundle. For example, a Health Care bundle might contain a Funding category for assemblies related to the management of hospital funds. All assemblies must be assigned to a category—they cannot be assigned to the root of a bundle.
Assemblies, bundles, and categories can be created and uploaded to the isee Exchange™ via the options on the Manage My Assemblies page. To learn more, visit our help pages, or take our assemblies tutorial.
Sim App (Sim)
An interface that allows users to interact with a model.
Sim apps allow users to interact with a model using buttons, sliders, knobs, tables, graphs, and storytelling. These interactions help users understand how parts of a system interact.
Interfaces are created by model authors in the Stella desktop software and can be uploaded to the isee Exchange™.
Model
A diagram that represents how elements in a system influence one another.
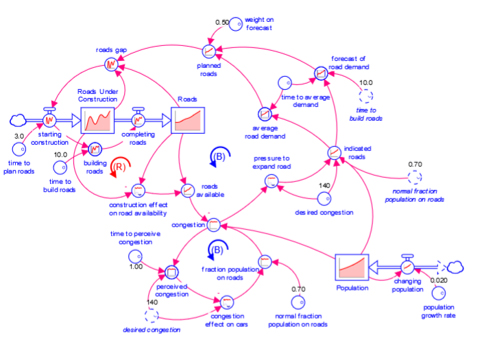
Models are mathematical representations of how elements in a system are connected and interact (e.g., ecosystems, organizations, supply chains). When running models on the isee Exchange™, results can be viewed in output devices like graphs and tables.
Models appear in the isee Exchange™ directory when authors upload them from the Stella® desktop software or create them with Stella Online™.
Causal Loop Diagram (CLD)
A map that represents the feedback structure of a system.
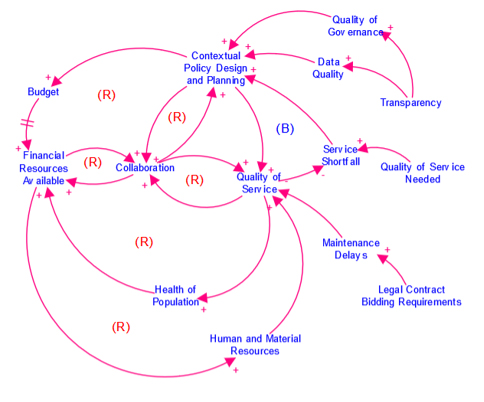
CLDs are high-level maps that represents the feedback structure of a system and easily communicate the essence of a model. They appear in the isee Exchange™ when authors upload them from the Stella desktop software or create them with Stella Online™.